Quick start
The following described:

Samples
It may be a sample of any mixture of small molecule compounds, such as animal metabolites, plant metabolites, drug samples, etc.
LC-MS/MS
For the workflow, SIRIUS has many restrictive Prerequisites that prevent its generalization to the analysis of any mass spectrometry data. Fortunately, your data as least will not be rejected by one vote as long as they meet the following two characteristics:
- High mass accuracy data
- Data-Dependent Acquisition (DDA) Mode.
Convert raw data
GNPS Server provides detailed instructions and services regarding data conversion. Click here.
If you are on Windows and have a lot of data to convert, then downloading locally and using MSconvert might be the best option. Click here
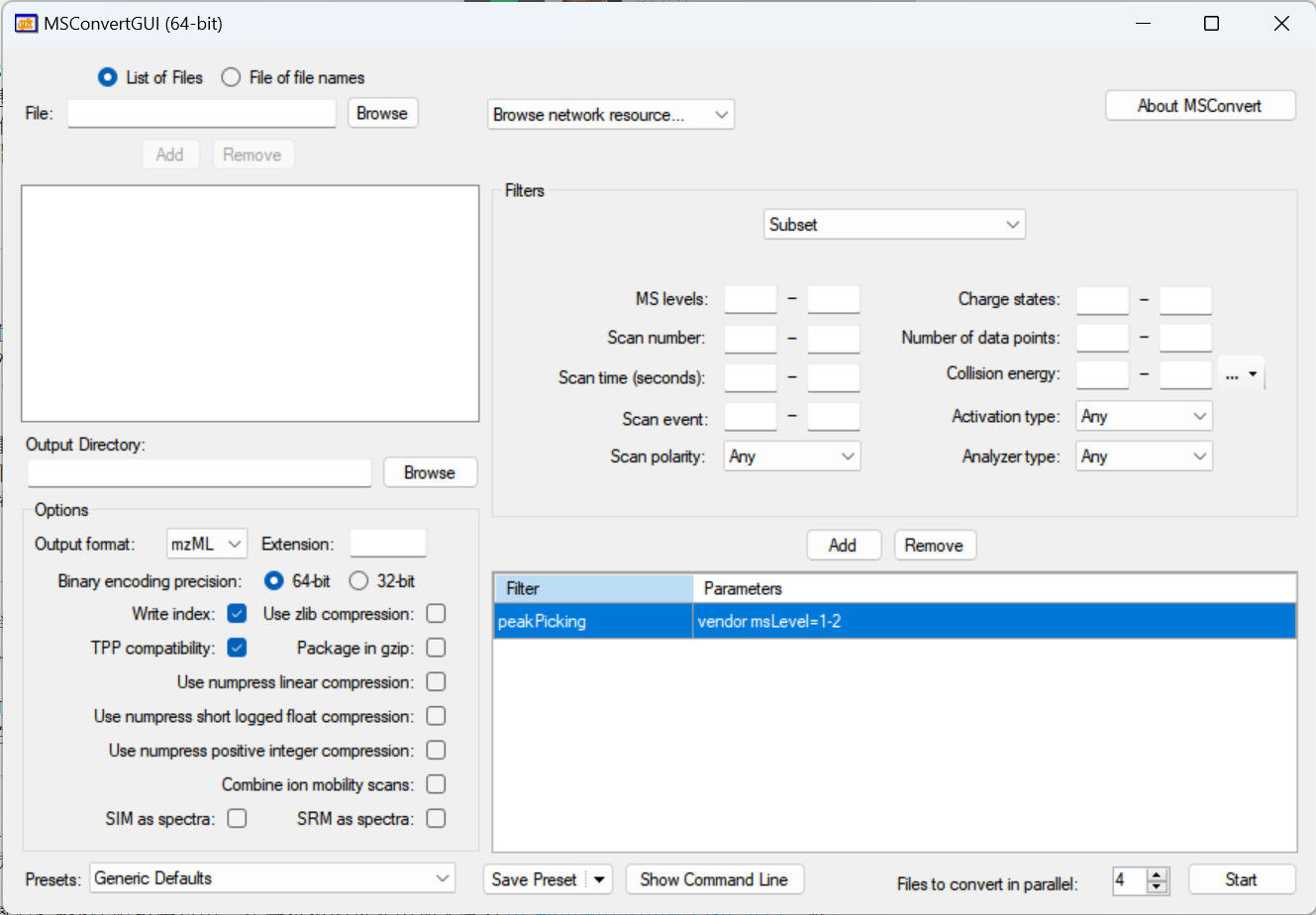
If you were not on Windows, installing the docker to perform MSconvert is
an option.
massconverter of TidyMass
provides a way in R to perform MSconvert with docker.
Feature detection

Feature Detection is a kind of algorithm for detecting peaks from EIC plots, and most mass spectrometry processing tools have a similar function. Users can implement this process with any tool, but to access the MCnebula workflow, .mgf (long list file containing MS/MS information) and .csv files (Feature quantification table) are required for output. The following are some examples of the four implementations of Feature Detection with output of .mgf files and .csv:
(Option 1) With MZmine
MZmine is a flexible deployment software for processing LC-MS data, providing a range of algorithms that can be freely combined to process LC-MS data according to user needs; its high degree of flexibility makes it potentially difficult to use.
MZmine has gone through several version changes and now MZmine3 is available. Unfortunately, our next example still uses version 2.53; since we did a test application with our Waters data when the original MZmine3 was released, only to find that errors occurred during data import, we are not sure if the latest version of MZmine3 has resolved this issue.
Download MZmine with version 2.53. Click here
The Guidance in GNPS provides a step by step description about MZmine for feature detection. Click here
Using batch mode of MZmine is the most convenient way to repeat a given process and parameters. We have provided some batch schema files (.xml) for MZmine2 for example. Click here
Note that different instruments and conditions may produce very different data, so it is a necessary step to pre-study your own data and customize the parameters when performing batch mode; ignore the parameter settings for the main steps, at least when applying these demo batch mode files, you will need to modify the input specified files and the output file names.
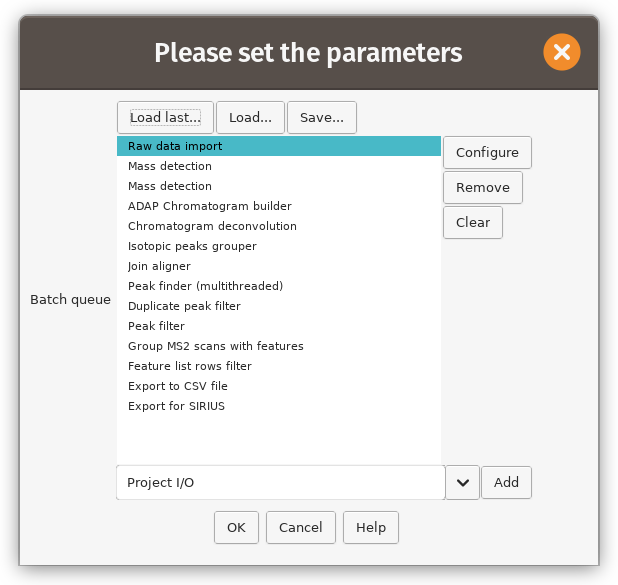
Demo XML for batch mode
(Option 2) With XCMS
Now, the R package MCnebula2 has a built-in module for Feature Detection, which uses pre-defined steps and parameters to quickly execute Feature Detection and get data (MS/MS lists and quantization tables) applicable to the MCnebula workflow. This module mainly contains a class called ‘mcmass’ and the method ‘run_lcms’. The pre-defined steps are encapsulated in an exemplary script based on the processing steps of FBMN: https://github.com/DorresteinLaboratory/XCMS3_FeatureBasedMN. Users need to change the input parameters and even add processing steps according to their data characteristics and research needs.
Install XCMS
If you do not already have xcms installed:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("xcms")
Build metadata for files
Hereinafter, file is the path where the mass spectrometry data you want to
process is located, sample is the alias you want to set for these data files,
and group is the group set-up for the files in study.
metadata <- data.frame(
file = c("EU-Pro1.mzML", "EU-Pro2.mzML", "EU-Raw1.mzML", "EU-Raw2.mzML"),
sample = c("pro-eu1", "pro-eu2", "raw-eu1", "raw-eu2"),
group = c("Pro", "pro", "raw", "raw")
)
Run LC-MS processing
Optional, you can set up parallel processing with BiocParallel.
set_biocParallel(4)
Create the mcmass object.
mcm <- new_mcmass(
metadata, snthresh = 5,
noise = 50000,
peakwidth = c(3, 30),
ppm = 20,
minFraction = 0.1
)
Checkout the steps:
detectFlow(mcm)
## layers of 5
## +++ layer 1 +++
## MSnbase::readMSData
## Args:
## files: character
## centroided.: logical
## mode: character
## pdata: NAnnotatedDataFrame
##
## +++ layer 2 +++
## xcms::findChromPeaks
## Args:
## object: toBeEval
## param: CentWaveParam
##
## +++ layer 3 +++
## xcms::adjustRtime
## Args:
## object: toBeEval
## param: ObiwarpParam
##
## +++ layer 4 +++
## xcms::groupChromPeaks
## Args:
## object: toBeEval
## param: PeakDensityParam
##
## +++ layer 5 +++
## xcms::fillChromPeaks
## Args:
## object: toBeEval
## param: ChromPeakAreaParam
Run processing and output .mgf:
mcm <- run_lcms(mcm)
anno <- run_export(mcm, saveMgf = "msms.mgf")
data.table::fwrite(features_quantification(mcm), "features.csv")
(Option 3) With OpenMS
Preparing…
(Option …) Any tools
You can use any tool to export a list containing MS/MS information and .csv. However, please Note that please convert the list of MS/MS information into a format that matches the SIRIUS input.
Run SIRIUS
SIRIUS is a java-based software framework for the analysis of LC-MS/MS data of metabolites and other “small molecules of biological interest” …
Sign up for use
Now, users must register to access from the SIRIUS network services (free for academic use) (sign up and login in the GUI).
Import .mgf
The .mgf file is obtained from the processing of the Feature Detection in the previous step, which records the basic information of the MS/MS. By dragging M into the GUI of SIRIUS, the data will be imported successfully. For command line mode, please refer to: Click here
The following is the content of a demo .mgf:
BEGIN IONS
FEATURE_ID=1
PEPMASS=532.30509842717
CHARGE=+1
MSLEVEL=1
532.30509842717 100
532.305646812001 100
533.309001652001 27.0393207318305
END IONS
BEGIN IONS
FEATURE_ID=1
PEPMASS=532.30509842717
CHARGE=+1
RTINSECONDS=
MSLEVEL=2
153.8885 148
181.8 74
334.8798 1881
360.7695 139
514.1035 120
451.17258 18.81
END IONS
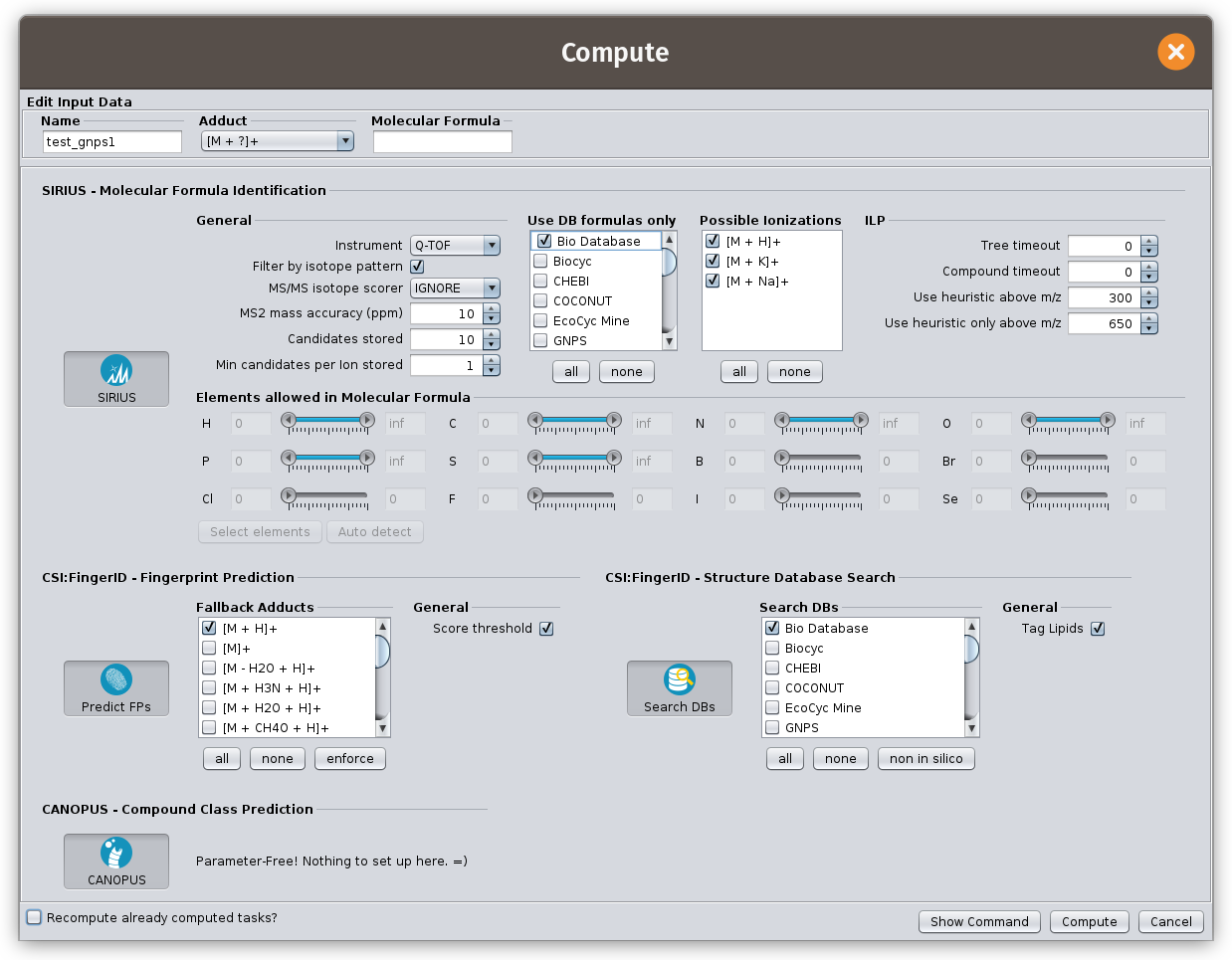
Activate modules
Activate SIRIUS, ZODIAC, CSI:FingerID and CANOPUS for computation. (If necessary, adjust the parameters)
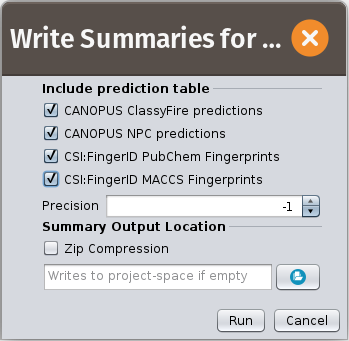
Write summaries
It may take hours or even an evening to calculate the LC-MS/MS data set containing thousands of Features. When it is over, write a summary.
MCnebula2
Now, let’s get started with the R package MCnebula2!
## The `path` is where your SIRIUS project saved.
path <- "."
mcn <- mcnebula()
mcn <- initialize_mcnebula(mcn, "sirius.v5", path)
<<< Workflow >>>